
Dušenova mišična distrofija (DMD) predstavlja najpogostejšo in najhujšo obliko dednih mišičnih bolezni pri otrocih. Njeni korenini segajo v bolezensko spremenjen genski zapis v genu DMD, ki se nahaja na X kromosomu. Ta genetska okvara vodi v progresivno atrofijo mišic, kar neizogibno vodi do popolne izgube samostojnosti in žal tudi do zgodnje smrtnosti. Ključni krivec za to degeneracijo je pomanjkanje distrofina, bistvene beljakovine citoskeleta, ki pri vretenčarjih igra ključno vlogo v membrani mišičnih vlaken, omogočajoč pravilno krčenje mišic.
Razumevanje Genetske Osnove in Mehanizmov Bolezni
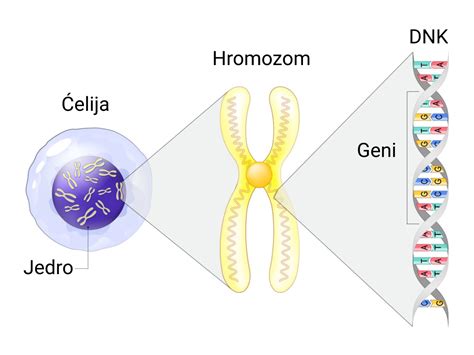
Mišične distrofije kot skupina dednih bolezni temeljijo na mutacijah genov, ki kodirajo beljakovine, ključne za normalno delovanje mišičnih celic. Pri vsaki specifični vrsti mišične distrofije so mutirani različni geni. Duchennova mišična distrofija, z mednarodno okrajšavo DMD (ORPHA: 98896), je X-vezana recesivna bolezen, ki primarno prizadene moške. Incidenca te bolezni je ocenjena na 1 na 3.300 novorojencev moškega spola. Prenaša se recesivno spolno vezano na kromosom X, kar pomeni, da moški, ki imajo le en X kromosom, izraziteje zbolijo, če je gen na tem kromosomu okvarjen. Ženske, ki imajo dva X kromosoma, običajno ne zbolijo, a lahko postanejo nosilke bolezni in prenesejo okvarjen gen na svoje potomce. Tveganje ponovitve bolezni pri moških potomcih je 50%.
Bolezenska okvara, ki povzroča DMD, je popolna odsotnost mišične beljakovine distrofina v skeletni in srčni mišici. Ta beljakovina je ključna za ohranjanje zgradbe mišičnih celic, natančneje za stabilnost sarkoleme, najmanjše funkcionalne enote mišice. Odsotnost distrofina vodi v napredujoč propad mišic. Gen DMD, ki se nahaja na lokaciji Xp21.2, kodira več različic zapisa, ključnih za sintezo distrofina.
Klinična Slika: Od Zgodnjih Znakov do Resnih Zapletov
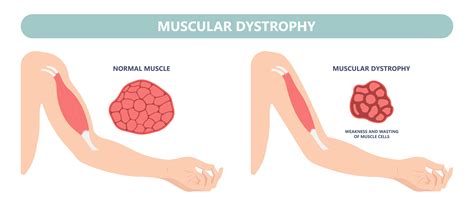
Klinična slika Duchennove mišične distrofije se običajno razvije v zgodnjem otroštvu, pogosto med tretjim in sedmim letom starosti. Ključni simptomi vključujejo progresivno mišično slabost, ki se najprej pojavi v proksimalnih delih spodnjih okončin in medenice. Z napredovanjem bolezni se slabost širi na zgornje ude, vrat in druge dele telesa. Značilni zgodnji znaki pri dečkih so lahko:
- Zaostajanje v motoričnem razvoju: Otroci lahko kasneje osvojijo nadzor nad glavo, sedenje, plazenje in samostojno vstajanje. Tek in skakanje sta pogosto nedosegljiva.
- Zibajoča hoja in Gowerjev manever: Bolezen se kaže z značilno zibajočo hojo. Gowerjev manever, pri katerem se otrok s tal dvigne s pomočjo opiranja rok na stegna, je pogost znak.
- Težave pri hoji po stopnicah in vstajanju: Vzpenjanje po stopnicah postane naporno, pogosti pa so tudi padci.
- Psevdohipertrofija meč: Kljub oslabelosti se lahko pojavi nenormalno povečanje mečnih mišic, ki pa je posledica nadomeščanja mišičnega tkiva z vezivom in maščevjem, kar pomeni, da te mišice niso močnejše.
- Zmanjšana vzdržljivost: Otroci imajo pogosto nizek nivo vzdržljivosti.
Z izgubo neodvisnosti pri gibanju, ki se pri nezdravljenih bolnikih pojavi v povprečju okoli 9,5 leta starosti (med 6. in 13. letom), se hitro razvijajo kontrakture sklepov in skoliotična deformacija hrbtenice. Pri 90% dečkov z DMD se pojavi tudi oslabelost srčne mišice, kar lahko povzroči težave z utripanjem srca, kar se lahko zazna na elektrokardiogramu. Poleg tega je prisotna lahko tudi neprogresivna slabša kognitivna funkcija, kar se lahko kaže v težavah s pomnjenjem.
Pri ženskah, ki so nosilke okvarjenega gena, se lahko bolezen pojavi kasneje, pogosto šele v odraslosti, s sliko blage do zmerne mišične šibkosti, ki je bolj proksimalna in nesimetrična.
Diagnostične Metode: Od Klinične Slike do Genetske Potrditve
Postavitev diagnoze mišične distrofije temelji na kombinaciji kliničnega pregleda, laboratorijskih testov in genetskega svetovanja. Klinični sum na DMD temelji na značilni klinični sliki, družinski anamnezi in izvidih laboratorijskih preiskav.
- Krvni testi: Pokažejo okvaro mišičnih celic. Raven kreatin-kinaze (CK) v serumu je pri DMD pogosto 50 do 200-krat višja od normalne, pri Beckerjevi mišični distrofiji pa 10- do 35-krat višja. Povišani so tudi encimi transaminaze (AST, ALT), ki uhajajo iz poškodovanih mišičnih celic.
- Mišična biopsija: Ta invazivna preiskava vključuje odvzem majhnega koščka mišice, ki se nato pregleda pod mikroskopom. Pri DMD biopsija pokaže propad mišičnih vlaken in odsotnost beljakovine distrofina.
- Genetsko testiranje: Analiza samega gena potrjuje prisotnost mutacij specifičnih genov in identificira specifično bolezen, ki povzroča mutacijo. V primeru DMD analiza DNK potrdi bolezensko različico v genu DMD, ki je lahko v obliki delecije s premikom bralnega okvirja, duplikacije ali nepomembne (nonsense) mutacije. Genetsko testiranje lahko predvidi tudi tveganje za razvoj bolezni. Za pričakovane matere lahko genetske študije, izvedene med nosečnostjo, zaznajo DMD z natančnostjo približno 95%.
Dedne genetske motnje | Genetika | Biologija | FuseSchool
Zdravljenje in Obvladovanje Bolezni: Celostni Pristop
Trenutno za Duchennovo mišično distrofijo ni zdravila, ki bi omogočalo popolno ozdravitev. Vendar pa obstajajo terapevtski pristopi, ki lahko pomagajo pri obvladovanju simptomov, upočasnitvi napredovanja bolezni in izboljšanju kakovosti življenja posameznikov. Zdravljenje DMD je multidisciplinarno in dolgotrajno, prilagojeno stopnji bolezni in funkcionalnim zmožnostim posameznika.
- Kortikosteroidi: Zdravila, kot so prednizon, prednizolon ali deflazakort, so se izkazala za učinkovita pri upočasnjevanju napredovanja bolezni. Delujejo tako, da stabilizirajo mišično membrano in pomagajo izboljšati mišično funkcijo. Uvedba kortikosteroidne terapije se pogosto odloči, ko otrokove motorične sposobnosti dosežejo plato, običajno med 4. in 8. letom starosti, pri čemer se individualno ocenjujejo funkcionalno stanje, starost in dejavniki tveganja za stranske učinke. Ob tem je ključno spremljanje in obvladovanje zapletov kortikosteroidne terapije, kot so prekomerna telesna teža, poškodbe želodčne sluznice (zahteva zdravljenje s H2-antagonisti), osteoporoza ter redno oftalmološko spremljanje za morebitno zdravljenje katarakte in glavkoma. Kortikosteroidno zdravljenje lahko upočasni propad mišic in podaljša obdobje samostojne mobilnosti za nekaj let.
- Fizioterapija: Redna telesna vadba je ključnega pomena, vendar mora biti skrbno prilagojena posamezniku, saj lahko pretirana ali visoko intenzivna vadba povzroči dodatno poškodbo mišic, medtem ko neaktivnost vodi v hitrejšo izgubo funkcije. Fizioterapevti priporočajo vaje za ohranjanje gibljivosti, krepitev mišic, izboljšanje drže in ravnotežja. Pasivno raztezanje ter nameščanje ortoz za gležnje in stopala preko noči lahko pomagajo zmanjševati kontrakture Ahilove tetive. Vadba mora biti primerno prilagojena in skrbno stopnjevana. Dihalni vaje izboljšujejo pljučno kapaciteto, kar je še posebej pomembno zaradi oslabelosti respiratornih mišic. Z vajami za povečanje obsega gibanja in raztezanja pod nadzorom fizioterapevta se ohranja večja prožnost sklepov. Aerobna vadba, kot je plavanje ali hoja, pomaga ohranjati moč, gibljivost in splošno zdravje. Fizioterapevti lahko uporabljajo dinamometer za merjenje mišične moči in spremljanje napredka ter ocenjujejo vzdržljivost pri hoji. Nadzorovana vadba pomaga ohranjati optimalen obseg gibljivosti sklepov in dobro mišično moč ter preprečuje atrofijo. Pod nadzorom fizioterapevta se izvaja tudi učenje pravilnega dihanja z izkašljevanjem. Fizioterapija na domu je lahko učinkovita in celostna rešitev.
- Ortopedski pripomočki: Opornice ali ortoze lahko pomagajo pri podpori sklepov in preprečujejo kontrakture ali deformacije. V primeru izjemno oslabljenih mišic se lahko poslužujete protez, vozičkov in drugih medicinskih pripomočkov, ki zagotavljajo oporo mišicam. Zaradi zmanjšane kostne gostote in povečanega tveganja za zlome se priporočajo fizioterapija, aerobna vadba ter zadostna količina beljakovin in kalcija v prehrani.
- Srčno zdravljenje: Redno spremljanje srčne morfologije in funkcije je potrebno za pravočasno uvedbo zdravljenja z ACE zaviralci, če pride do prizadetosti srčne mišice.
- Kirurško zdravljenje: V nekaterih primerih je za korekcijo skolioze potrebno kirurško zdravljenje.

Nove Terapevtske Potenciali: Proteomika in Genska Terapija
Napredek v raziskavah prinaša nova upanja za zdravljenje Duchennove mišične distrofije. Nedavno odkritje, da celična komunikacija med mišicami in limfnim sistemom igra vlogo pri DMD, odpira vrata razvoju novih načinov zdravljenja.
- Proteomika: Raziskovalci z Univerze v Bonnu so z uporabo proteomike pri miših odkrili, kako skeletne mišice in vranica vplivata druga na drugo zaradi pomanjkanja distrofina. Proteomika, kot zanesljiva in učinkovita analitična metoda, omogoča prepoznavanje beljakovin, značilnih za različne bolezni, in prinaša informacije o poteku bolezni ter možnih terapevtskih ciljih. Obolele miši so pokazale številne spremembe v proteomskem "podpisu" v primerjavi z zdravo kontrolno skupino. Odkrili so tudi krajšo obliko distrofina (DP71), ki se sintetizira v vranici in na katero očitno ne vpliva pomanjkanje dolge oblike. Študija potrjuje, da izguba dolge oblike distrofina v skeletnih mišicah povzroča sekundarne učinke na limfni sistem, kar poudarja pomen razumevanja vnetnih procesov, ki so pomembna značilnost degeneracije mišičnih vlaken.
- Genska terapija in nova zdravila:
- Ataluren: To zdravilo, ki je že priznano v nekaterih državah, učinkuje pri manjši skupini bolnikov (približno 13%) z specifično gensko napako - nepomembno (nonsense) bolezensko spremembo gena za distrofin. Ataluren terapevtsko posreduje pri prepisovanju in nastajanju beljakovine distrofina tako, da premosti napako v genskem zapisu (stop codon read-through), kar poveča delež aktivnega distrofina in upočasni potek bolezni.
- Eteplirsen: V ZDA že registrirano zdravilo, ki s tehniko "ekson skipinga" (exon skipping) pomaga preskočiti napako v dednem zapisu za distrofin. Eteplirsen je učinkovit pri premostitvi eksona 51, kjer je najpogostejša napaka pri DMD. Omogoča, da se genski zapis za distrofin prepisuje dalje preko napake, kar povzroči nastanek nekoliko krajše, a še vedno delujoče beljakovine distrofina.
- Givinostat (Duvyzat): To je zaviralec HDAC (histonskih deacetilaz), encimov, ki sodelujejo pri uravnavanju izražanja genov. Givinostat lahko zmanjša regeneracijo mišic pri Duchennovi mišični distrofiji. Evropska unija je nedavno odobrila Duvyzat za zdravljenje Duchennove mišične distrofije pri otrocih, starejših od 6 let, ki še lahko hodijo. Odobritev temelji na pozitivnih rezultatih kliničnih študij, ki kažejo na upočasnitev napredovanja bolezni.
- Vamorolon (Agamree): Nov steroid, ki se uporablja za zdravljenje Duchennove mišične distrofije.
Raziskovalni projekti za nove oblike zdravljenja DMD so v polnem teku, kar daje upanje na izboljšanje prognoze in kakovosti življenja bolnikov v prihodnosti.
Prognoza in Življenjska Doba
Duchennova mišična distrofija ima kljub napredkom v zdravljenju še vedno slabo prognozo, pričakovana življenjska doba pa je znatno skrajšana. Brez ustreznega zdravljenja, večina otrok z DMD umre pred 20. letom starosti zaradi napredujoče oslabelosti, respiratornih zapletov in srčnih težav. Vendar pa lahko sodobni terapevtski pristopi, vključno s kortikosteroidi, fizioterapijo in novimi zdravili, podaljšajo življenjsko dobo in izboljšajo njeno kakovost.
Dedne genetske motnje | Genetika | Biologija | FuseSchool
Življenje s kronično boleznijo, kot je mišična distrofija, prinaša tudi čustvene izzive. Psihološka podpora, bodisi individualna ali skupinska terapija, je lahko ključnega pomena za bolnike in njihove svojce pri obvladovanju teh izzivov.
tags: #misicna #distrofija #pri #dojencku